
Cystic Fibrosis
Introduction
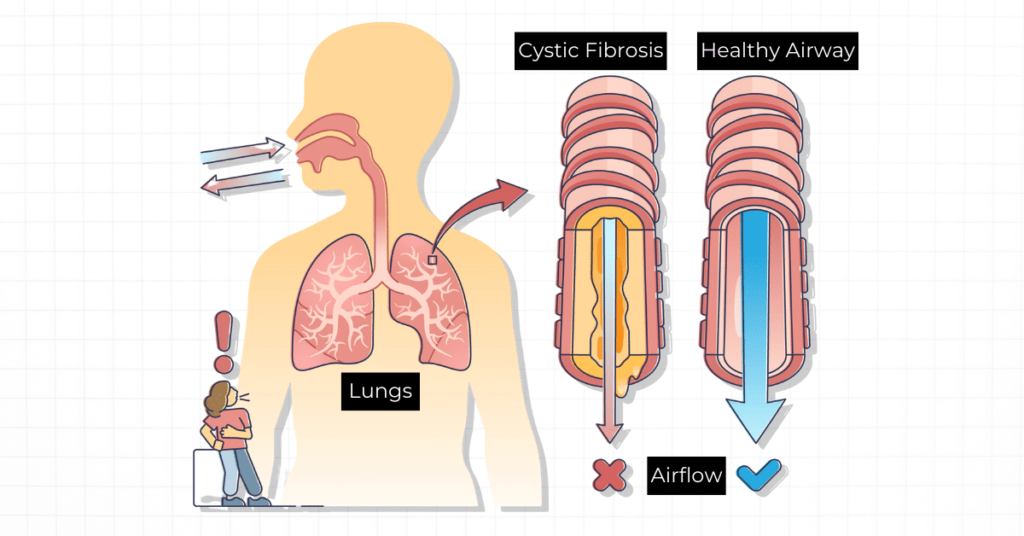
Cystic fibrosis (CF) is a genetic disorder that affects various organ systems, primarily the lungs and digestive system. It is a chronic and progressive condition caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which regulates the movement of salt and water across cell membranes. This genetic defect leads to the buildup of thick, sticky mucus in various organs, resulting in a range of debilitating symptoms and complications.
Causes
It is an inherited disease caused by mutations in the CFTR gene. This gene provides instructions for producing the CFTR protein, which plays a crucial role in regulating the movement of salt and water across cell membranes. Mutations in the CFTR gene disrupt the normal function of this protein, leading to the production of abnormally thick and sticky mucus.
Risk Factors
CF is an autosomal recessive disorder, meaning that an individual must inherit two faulty CFTR genes, one from each parent, to develop the condition. The risk factors for having a child with cystic fibrosis include:
1. Family history: If both parents are carriers of a CFTR gene mutation, they have a 25% chance of having a child with cystic fibrosis with each pregnancy.
2. Ethnicity: CF is more common in certain ethnic groups, such as Caucasians and Ashkenazi Jews.
3. Consanguinity: The risk of having a child with cystic fibrosis is higher if the parents are closely related, as they may share a common ancestry and carry the same CFTR gene mutation.
Symptoms
1. Respiratory symptoms: Persistent cough, wheezing, shortness of breath, and recurrent lung infections (such as pneumonia and bronchitis) due to the buildup of thick mucus in the lungs.
2. Digestive symptoms: Poor absorption of nutrients, greasy and foul-smelling stools, abdominal pain, and poor growth or weight gain due to pancreatic insufficiency (lack of digestive enzymes).
3. Salty skin: Individuals with cystic fibrosis may have salty-tasting skin due to the increased concentration of salt in their sweat.
4. Infertility: Men with cystic fibrosis may experience infertility due to the absence of the vas deferens (the tube that carries sperm from the testicles).
Complications
It’s can lead to various complications, many of which are related to the buildup of thick mucus in various organs. These complications include:
1. Lung damage and respiratory failure: The accumulation of mucus in the lungs creates an environment conducive to bacterial growth, leading to chronic lung infections, inflammation, and progressive lung damage, which can ultimately result in respiratory failure.
2. Malnutrition and growth deficiencies: The impaired absorption of nutrients due to pancreatic insufficiency can lead to malnutrition, vitamin deficiencies, and stunted growth in children with cystic fibrosis.
3. Diabetes: Individuals with cystic fibrosis have an increased risk of developing cystic fibrosis-related diabetes (CFRD), a distinct form of diabetes caused by scarring and damage to the pancreas.
4. Liver disease: The buildup of mucus in the liver can lead to biliary cirrhosis, a condition characterized by scarring and damage to the liver.
5. Infertility: While women with cystic fibrosis can become pregnant, men with the condition are often infertile due to the absence of the vas deferens.
Differential Diagnosis
CF shares some symptoms with other respiratory and digestive conditions, and it is essential to differentiate it from these conditions for proper diagnosis and treatment. The differential diagnosis for CF may include:
1. Asthma: Both cystic fibrosis and asthma can cause wheezing and shortness of breath, but cystic fibrosis typically involves additional symptoms like chronic cough and digestive issues.
2. Chronic bronchitis: Chronic bronchitis can cause persistent cough and mucus production, similar to cystic fibrosis, but it does not involve the digestive and other systemic manifestations of cystic fibrosis.
3. Bronchiectasis: This condition, characterized by abnormally dilated and damaged bronchial tubes, can cause symptoms similar to cystic fibrosis, but it is not an inherited disorder and does not involve digestive issues.
4. Celiac disease: Like cystic fibrosis, celiac disease can cause malabsorption of nutrients and poor growth, but it is an autoimmune disorder triggered by gluten exposure and does not involve respiratory symptoms.
5. Primary ciliary dyskinesia: This rare genetic disorder affects the movement of cilia (tiny hair-like structures) in the respiratory tract, leading to recurrent respiratory infections and bronchiectasis, but without the digestive and other systemic manifestations of cystic fibrosis.
Preventions
It is an inherited genetic disorder, and there is no known way to prevent its occurrence. However, certain measures can be taken to manage the condition and reduce the risk of complications:
1. Genetic counseling and prenatal testing: Couples with a family history of cystic fibrosis or those from high-risk ethnic groups can undergo genetic counseling and prenatal testing to assess the risk of having a child with the condition.
2. Newborn screening: Many countries have implemented newborn screening programs to identify infants with cystic fibrosis shortly after birth, allowing for early intervention and treatment.
3. Avoiding environmental triggers: Individuals with cystic fibrosis should avoid exposure to triggers that can exacerbate respiratory symptoms, such as cigarette smoke, air pollution, and respiratory infections.
4. Regular exercise and physical therapy: Engaging in regular exercise and chest physical therapy can help improve lung function and clear mucus from the airways.
Diagnosis
1. Newborn screening: In many countries, newborns are screened for cystic fibrosis through a blood test that measures the levels of certain markers associated with the condition.
2. Sweat test: The sweat test is the standard diagnostic test for cystic fibrosis. It measures the concentration of chloride (salt) in the sweat, which is typically elevated in individuals with the condition.
3. Genetic testing: Genetic testing can identify mutations in the CFTR gene and confirm the diagnosis of cystic fibrosis.
4. Clinical evaluation: A thorough medical history, physical examination, and assessment of symptoms, such as respiratory and digestive issues, can provide valuable information in diagnosing cystic fibrosis.
Treatments
It is a chronic and progressive condition that requires a multidisciplinary approach to treatment. The primary goals of treatment are to manage symptoms, reduce complications, and improve the overall quality of life. Treatment strategies may include:
1. Airway clearance techniques: Chest physical therapy, including percussion and vibration techniques, as well as the use of devices like vests or masks that apply high-frequency vibrations, can help loosen and remove mucus from the lungs.
2. Medications: Various medications may be prescribed to manage symptoms and complications, such as bronchodilators to improve airway function, antibiotics to treat lung infections, and pancreatic enzyme supplements to aid in digestion.
3. Nutritional support: A well-balanced, high-calorie, and high-fat diet, along with vitamin and mineral supplements, is crucial to ensure adequate growth and nutrition.
4. Exercise and physical activity: Regular exercise and physical activity can help improve lung function, promote airway clearance, and maintain overall fitness.
5. Lung transplantation: In severe cases of advanced lung disease, a lung transplant may be considered as a last resort for individuals with cystic fibrosis.
6. Gene therapy and CFTR modulators: Ongoing research is exploring the potential of gene therapy and CFTR modulators (medications that target the underlying genetic defect) to correct or compensate for the CFTR gene mutations.
Important Key Points
- Cystic fibrosis is a genetic disorder caused by mutations in the CFTR gene, leading to the production of thick, sticky mucus in various organs.
- It primarily affects the lungs and digestive system, causing respiratory and digestive issues.
- Risk factors include family history, being from certain ethnic groups (Caucasians, Ashkenazi Jews), and consanguinity (parents being closely related).
- Symptoms include persistent cough, wheezing, shortness of breath, poor growth/weight gain, greasy stools, salty skin, and infertility in males.
- Complications include lung damage, respiratory failure, malnutrition, diabetes, liver disease, and infertility.
- Differential diagnosis includes asthma, chronic bronchitis, bronchiectasis, celiac disease, and primary ciliary dyskinesia.
- Prevention involves genetic counseling, prenatal testing, newborn screening, and avoiding environmental triggers.
- Diagnosis is made through newborn screening, sweat test, genetic testing, and clinical evaluation.
- Treatment aims to manage symptoms, reduce complications, and improve quality of life through airway clearance techniques, medications, nutritional support, exercise, and, in severe cases, lung transplantation.
- Gene therapy and CFTR modulators are areas of ongoing research for potential treatments targeting the underlying genetic defect.
- A multidisciplinary team approach is essential for comprehensive management of cystic fibrosis.
It is important for individuals with cystic fibrosis to work closely with a multidisciplinary team of healthcare professionals, including pulmonologists, gastroenterologists, nutritionists, physical therapists, and genetic counselors, to develop a comprehensive and personalized treatment plan.
Note: This is a general overview, and it’s always best to consult with a healthcare professional for personalized medical advice and treatment.